Audit-Ready Clinical Trial Documents.
First Draft in One Business Day.
Skaldi generates source-traceable first drafts of Investigator's Brochures, Protocols, CSRs, and 5 more document types. Your team reviews and refines — instead of starting from blank pages.
Three inputs. One platform. Complete documentation suite.
Built by medical writers with 15+ years in pharma
Novartis · Bayer · Sandoz · BIOCAD
The Problem: Clinical Documentation Still Runs on Manual Labor
Weeks of Manual Drafting
A single Investigator's Brochure takes 4-8 weeks and $30-50K. A Protocol — another 2-4 weeks. Multiply by every document in your submission package.
Cross-Document Contradictions
Objectives in the Protocol don't match the SAP. Endpoints in the IB conflict with the CSR. Every inconsistency triggers audit findings and revision cycles.
Manual Literature Search
40+ hours per document searching PubMed, FDA labels, and EPARs. And still missing critical references that reviewers will ask about.
Eight Document Types. One Workflow.
Every document enriched from regulatory databases, cross-referenced for consistency, and validated against ICH and FDA requirements.
Investigator's Brochure
Comprehensive compound profile with full evidence mapping from FDA, PubMed, and DrugBank.
See Sample →Clinical Protocol
Study design, endpoints, procedures, and schedules of assessments — structured and ICH E6-aligned.
Clinical Study Report
ICH E3-compliant report generation with complete efficacy, safety, and statistical sections.
Statistical Analysis Plan
Pre-specified analyses aligned with protocol endpoints and regulatory expectations.
Informed Consent Form
Patient-facing language at appropriate reading level. IRB-ready formatting.
Study Synopsis
Concise study summary for regulatory submissions and internal review.
Summary of Product Characteristics
EU SmPC with standardized sections, safety data, and prescribing information.
Case Report Form
Data collection instruments aligned with protocol visits and procedures.
How Skaldi Works
Four simple steps from study design to regulatory-ready documentation
Provide Three Things
Enter:
- Drug (INN or molecule)
- Indication
- Primary objectives
Skaldi automatically pulls study patterns, endpoints, procedures, and evidence.
AI Research & Evidence Collection
Skaldi scans PubMed, FDA labels, EMA EPARs, ClinicalTrials.gov, and guideline libraries to assemble a validated evidence foundation for your document.
Automated Drafting & Validation
A full document draft is generated with continuous validation against:
- ICH E6(R3), E3, E7, E8, and E9
- FDA 21 CFR Parts 11, 50, 56, and 312
- Internal consistency rules
- Cross-document alignment
Validation runs continuously as the document is generated.
Review & Export
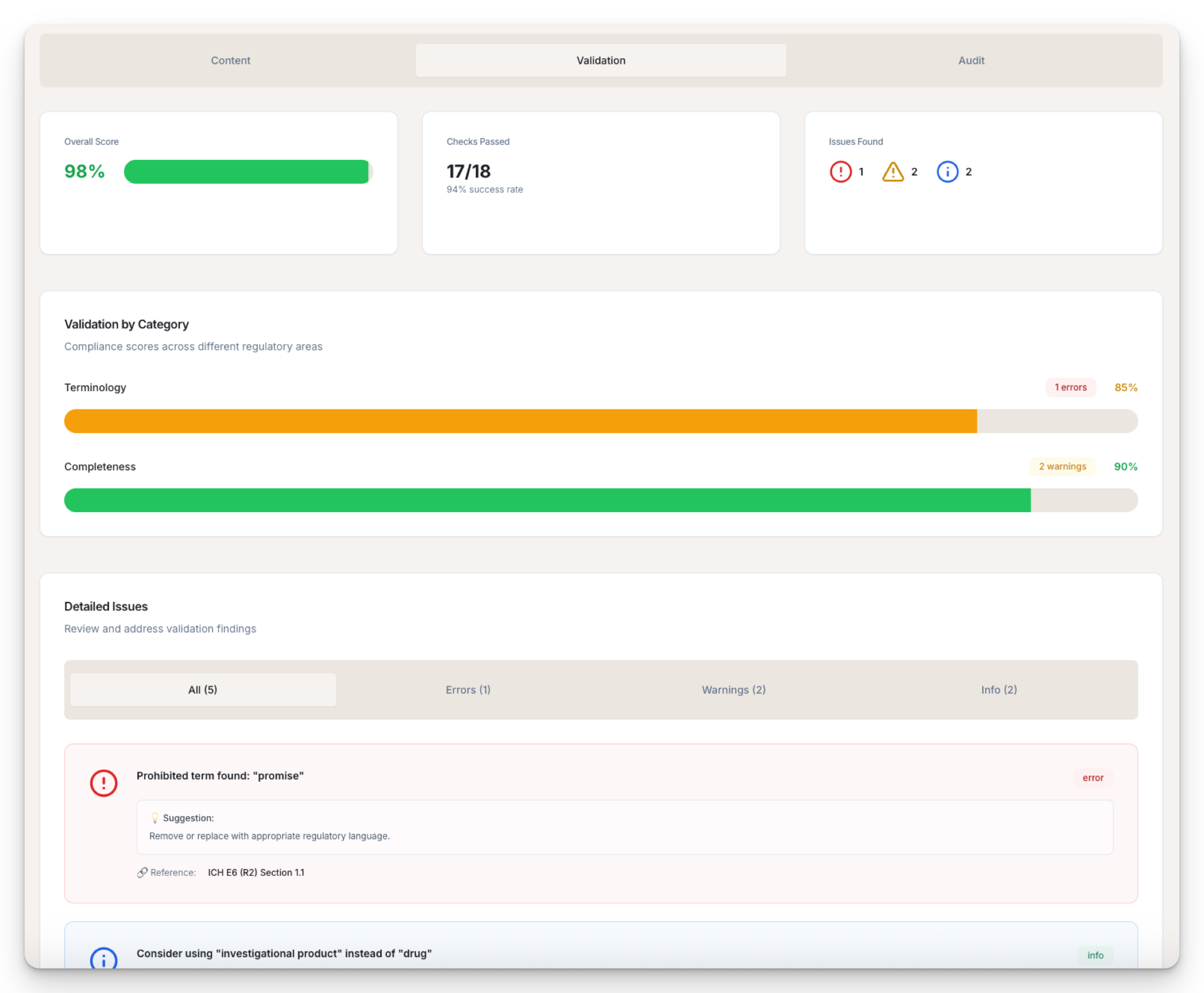
Compare versions, apply edits, comment, and export audit-ready DOCX or PDF with full version control and audit trail.
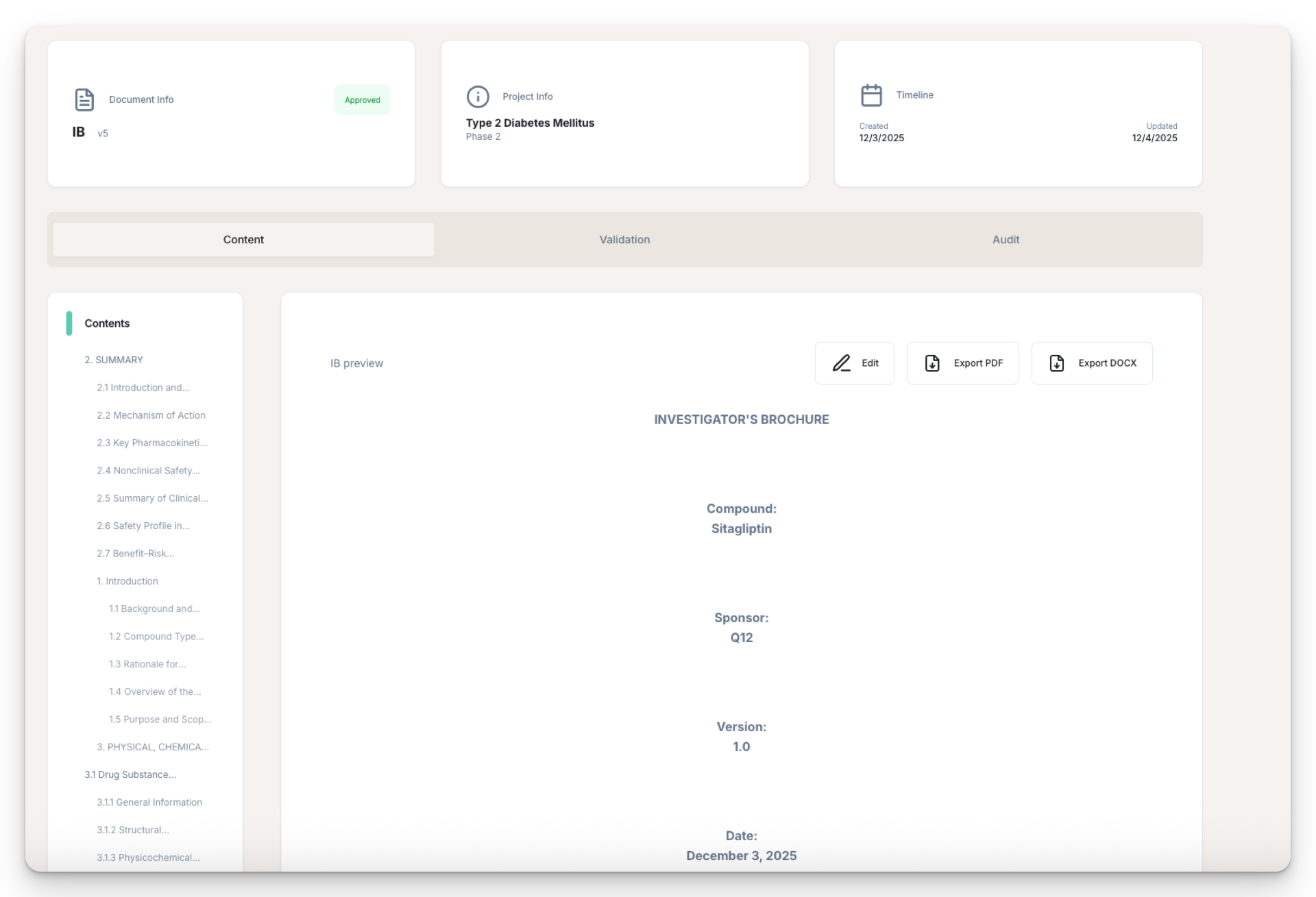
Investigator's Brochure: First Draft, Source-Mapped, in One Business Day
This IB for Adalimumab (Biosimilar to HUMIRA®, 351(k) pathway) was generated from three inputs. 65 pages. 10 sections. Full audit trail.
Sample Investigator's Brochure
Adalimumab · 351(k) Biosimilar Pathway
Title Page
Confidential
Investigator's Brochure
Adalimumab-atto
Biosimilar to HUMIRA® — Solution for Subcutaneous Injection
351(k) Biosimilar Pathway
Edition: 2 | Date: April 2, 2026
Generated by Skaldi AI Platform
3. Summary
Adalimumab is a recombinant human IgG1 monoclonal antibody that binds specifically to tumor necrosis factor-alpha (TNF-α), neutralizing its biological activity. The investigational product is a proposed biosimilar to the reference product HUMIRA®.
The product is being developed under Section 351(k) of the Public Health Service Act. Analytical similarity has been demonstrated through extensive physicochemical and functional characterization comparing the investigational product to HUMIRA®.
Clinical biosimilarity is being evaluated in patients with rheumatoid arthritis, the most sensitive therapeutic indication for anti-TNF-α monoclonal antibodies. The totality of evidence supports extrapolation to all approved HUMIRA® indications.
5. Analytical Similarity Assessment
Comprehensive analytical similarity was evaluated across multiple quality attributes. Results demonstrate high similarity between the proposed biosimilar and HUMIRA® across all critical quality attributes.
Table 3-1. Analytical Similarity Summary
9. Investigator Guidance
Safety monitoring should follow the schedule below. Investigators should pay particular attention to injection site reactions and immunogenicity signals.
Table 9-6. Safety Monitoring Schedule
10. References
All claims are source-traceable to regulatory databases, peer-reviewed literature, and clinical trial registries.
- FDA. HUMIRA® (adalimumab) Prescribing Information. Reference ID: 4753092. NDA 125057 ↗
- Kaur P, et al. "Analytical similarity assessment of proposed biosimilar adalimumab to HUMIRA®." BioDrugs. 2021;35(4):459-472. PMID: 34264503 ↗
- Cohen SB, et al. "Switching from adalimumab to biosimilar SB5 in patients with RA: 52-week efficacy, safety, and immunogenicity." Ann Rheum Dis. 2018;77(Suppl 2):476. NCT02167139 ↗
- FDA Guidance for Industry. "Scientific Considerations in Demonstrating Biosimilarity to a Reference Product." April 2015. FDA Guidance ↗
- Weinblatt ME, et al. "Phase III randomized study of SB5 vs reference adalimumab in RA." Ann Rheum Dis. 2018;77(2):234-240. NCT02593434 ↗
We showcase the Investigator's Brochure because it's our most complex document type. If Skaldi delivers a source-mapped IB first draft in one business day, every other document type follows the same workflow.
Clinically Sound Study Design
Schedules of Assessments are generated and structured according to study phase, indication, and regulatory expectations — reducing inconsistencies and downstream amendments.

Enterprise-Grade Security and Compliance Foundation
Built on certified infrastructure with regulatory alignment at every level
Hosting and Infrastructure
Skaldi is hosted on Microsoft Azure, leveraging SOC 2 and ISO 27001 certified data centers with enterprise-grade physical and network security.
Data Protection
All data is encrypted in transit and at rest. Access is strictly controlled via role-based permissions and isolated per customer.
Compliance Alignment
Built on Azure services designed to meet GDPR and HIPAA requirements. SOC 2 Type II and ISO 27001 certification in progress.
Audit Readiness
The platform includes versioning, access logs, and audit trails to support QA and regulatory review workflows.
Regulatory Standards Supported
21 CFR Parts 11, 50, 56, and 312
European Medicines Agency Guidelines
E6(R3), E3, E7, E8, and E9
EU Data Protection
US Health Data Privacy
Built-In Regulatory Validation
Every document is continuously validated against ICH and FDA requirements. Skaldi flags prohibited terminology, missing sections, and logical gaps before QA or regulatory review.
Manual Process vs. Skaldi
| Manual | Skaldi | |
|---|---|---|
| Investigator's Brochure | 4-8 weeks, $30-50K | Under 24 hours (first draft) |
| Clinical Protocol | 2-4 weeks, $15-25K | Under 24 hours (first draft) |
| Clinical Study Report | 6-12 weeks, $50-80K | Under 48 hours (first draft) |
| Source traceability | Manual footnoting | Automatic, every claim |
| Cross-document consistency | Manual QC review | Built-in validation engine |
| Literature search | 40+ hours per document | Automatic enrichment |
Investigator's Brochure
Manual
4-8 weeks, $30-50K
Skaldi
Under 24 hours (first draft)
Clinical Protocol
Manual
2-4 weeks, $15-25K
Skaldi
Under 24 hours (first draft)
Clinical Study Report
Manual
6-12 weeks, $50-80K
Skaldi
Under 48 hours (first draft)
Source traceability
Manual
Manual footnoting
Skaldi
Automatic, every claim
Cross-document consistency
Manual
Manual QC review
Skaldi
Built-in validation engine
Literature search
Manual
40+ hours per document
Skaldi
Automatic enrichment
Estimates based on industry averages for outsourced medical writing. Skaldi times are for first draft generation. Source-traceable outputs require human medical writer review before regulatory use.
Built for Clinical Research Professionals
Whether you're a biotech, CRO, or pharmaceutical company, Skaldi adapts to your needs
Biotechs
Enterprise-grade documents without an enterprise team.
You're running lean and moving fast toward IND or Phase 2. Skaldi gives you the documentation quality of a top-10 pharma without hiring a medical writing department.
- Full documentation suite from day one
- Regulatory expertise built into every draft
CROs
Scale medical writing capacity without scaling headcount.
Handle more studies simultaneously with the same team. Skaldi generates first drafts, your medical writers review and refine.
- 95% reduction in first-draft turnaround
- Cross-document consistency across all studies
- Volume pricing for multi-study workflows
Pharma
Accelerate study startup. Cut documentation costs.
Reduce time from study design to regulatory submission. Maintain consistent quality across your clinical portfolio.
- Faster IND and CTA submissions
- Consistent quality across all trials
- Full audit trail for regulatory review
Submission-Ready Documents
Review, edit, version, and export audit-ready documents in DOCX or PDF — fully aligned with regulatory structure and clinical best practices.
Get Started
Pricing tailored to your stage and program scope. Pilot engagements available for clinical-stage biotech evaluating Skaldi for an upcoming IB, Protocol, or regulatory submission.
Mutual NDA executed before kickoff. Hosted on Microsoft Azure (SOC 2 / ISO 27001 certified infrastructure). GDPR and HIPAA aligned.
From the Clinical Docs Library
Expert guides on clinical trial documentation
Medical Writing for Clinical Trials
Medical writing fundamentals for clinical trials — document types, regulatory standards, quality principles, and career guidance for writers.
Read
Clinical Trial Protocol Template
Clinical trial protocol template with all ICH E6(R2) required sections — study design, objectives, eligibility, procedures, and statistics.
Read
FDA 21 CFR Part 11 Compliance Guide
Step-by-step FDA 21 CFR Part 11 compliance guide — system validation, electronic signatures, audit trails, and common compliance gaps.
ReadSee what Skaldi can do for your compound.
Free 5-page IB Section 5 (Effects in Humans) preview on any biotech compound. Source-mapped, public-data sourced. Delivered within 24 hours of request, via secure link.